|
|
||
|
A Mystery Unfolds Healthy proteins misfold into potentially deadly prions
|
|||||||
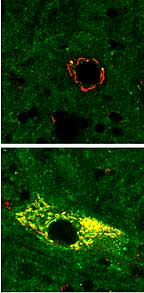
Prion diseases eat away at the brain. At top, a normal neuron, and below, an infected neuron with an accumulation of the abnormal, scrapie form of the prion protein. Scrapie is a fatal disease affecting sheep.
Humans get prion disorders from inherited mutations, through contami-nation during a medical procedure or, in very rare instances, from consumption of infected animals.
Like any genuinely new area of research, prions seem to have a habit of throwing unexpected surprises at scientists.
Connecting prion disease to other more common disorders, such as Alzheimer’s
| FOR RESEARCHERS LIKE David A. Harris, MD, PhD, the long, slow exit from the twilight zone is all but over. Harris has been studying prions, a new kind of infectious agent thought to be at the heart of several rare neurodegenerative disorders that devastate the brains of humans, cows and sheep. Prions are weird — unlike any other infectious agent ever identified before. Harris, professor of cell biology and physiology, remembers a time when describing his research sometimes gave him the impression other scientists thought he had “gone to outer space” or was working on “black magic.” Seven years after the Nobel Prize went to a prion researcher, Harris admits that an ironclad proof of prion theory has yet to be produced. But the skeptics are finding it harder and harder to make their case, and Harris now has a colleague in prion research in his own department, Heather L. True-Krob, PhD, assistant professor of cell biology and physiology. Harris and True-Krob are gathering new insights into how prions form and cause disease, and as they do, tantalizing hints are starting to emerge that prions may be connected to a much wider range of biological phenomena than the rare brain disorders that first led to their discovery.
Heather L. True-Krob, PhD, and David A. Harris, MD, PhD Until prions came along, infectious agents always contained some type of genetic material. That material carried the linchpin of the infection cycle: instructions for hijacking host cells to produce new copies of the infectious agent and begin the cycle anew. Not so the prion — it consists entirely of a misfolded protein. The prion perpetuates itself by influencing nearby normal copies of the same protein, somehow increasing the chances they will misfold and become prions. In cows with mad cow disease, sheep with scrapie, and humans with Creutzfeldt-Jakob disease, this causes a chain reaction that leaves the brain a spongy, hole-filled mess. Humans get prion disorders from inherited mutations, through contamination during a medical procedure or, in very rare instances, from consumption of infected animals. In addition, some “spontaneous” cases of human prion disease currently can’t be tracked to any genetic or environmental cause. The disorders have no treatment and are fatal in months to several years. The first part of prion theory, the idea that a change in folding can radically change a protein’s properties, is well-established biological fact. Proteins are long, complex chains, and as those chains fold up, they form specialized structures that can perform various functions. Rearranging the way a protein folds can eliminate those structures, create new structures, or change their accessibility. The process is roughly comparable to a Swiss Army knife: fold the protein in one configuration, and the can opener sticks out and can be used; fold it into another, and the can opener vanishes, a screwdriver sticks out, and the protein has suddenly become a tool used for a very different purposes. Much of the resistance among scientists to accepting prions springs from the second part of prion theory: the idea that interaction with a misfolded protein can cause another copy of the same protein to become badly folded. The details of how this unprecedented conversion takes place are still a mystery. “ The problem is that no one knows the exact three-dimensional structure of the prion,” Harris explains. “We know the normal structure of the protein that becomes a prion, but not the structure of the prion itself, and that’s left the process by which prions spread a kind of black box.” The normal function of the protein that becomes a prion also remains a mystery. Scientists named the protein PrP: The normally folded copy is referred to as PrPC (for cellular PrP), while the prion form is known as PrPSc (for scrapie PrP). Recent evidence even has scientists questioning one of their most basic assumptions about prions: the idea that PrPSc is the form of the prion protein that kills brain cells. Studies by Harris have shown that transgenic mice with a mutant form of PrP prone to forming prions will get symptoms like those in human prion disorders, but the disease is not infectious to other animals. “ In terms of the different forms of PrP, we have early evidence that what’s needed to kill a neuron may be different from what’s needed to pass on an infection,” Harris notes. Like any genuinely new area of research, prions seem to have a habit of throwing unexpected surprises at scientists. One such surprise has actually boosted acceptance of prions among the research community: the identification of prions in yeast cells. True-Krob specializes in the study of yeast prions, which don’t affect humans and other mammals but have similar structural elements. Yeast prions spread the same way as mammalian prions, with proximity to misfolded copies of the yeast prion protein, Sup35, somehow increasing the chances that normal copies of the same protein will become prions. During her postdoctoral studies, True-Krob led a project that uncovered another major prion surprise: a positive role for yeast prions. Sup35 normally helps yeast read protein-building instructions from its DNA, a process called translation. True-Krob showed that the prion form of Sup35 disrupted this process. As a result, new material was added to proteins. The switch to prion-prone Sup35, which occurs spontaneously about once in every million generations of yeast, often has harmful effects. But in about 20 percent of test cases, the disruptions gave the yeast a survival advantage in an environment in which temperature, the availability of food or other factors had changed. “This system is advantageous for the yeast because they have a way of turning prions on and off,” True-Krob notes. “And that gives us hope that what we learn from yeast may help us find a way to turn prions off in humans.” Working with prions in yeast lets True-Krob conduct studies that would be prohibitively complex or even impossible in mammalian cells. She can simultaneously expose many different yeast cell lines to a wide range of environmental conditions and genetic variables and see how these factors influence the likelihood that prions will form. True-Krob is active in the search for additional yeast prions, which has netted a second yeast prion also linked to the translation of information in DNA. “ People have speculated that there may be up to 100 different prions in yeast,” True-Krob says. “What we learn in yeast will help us search for prions in other systems.” Harris, who studies mammalian prions, describes his lab’s interests as the molecular and cellular biology of the prion protein: What do both forms of PrP do in the nerve cell, where do they do it, and what do they interact with? Harris conducts the bulk of his research in approximately 50 lines of mice genetically modified to produce prions and symptoms similar to human prion diseases. In recent years, they’ve produced important clues about what PrPC and PrPSc may be doing in the brain. Work in mouse models has shown that PrP scrapie builds up in clumps in the brain similar to those seen in more common neurodegenerative disorders like Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Another similarity to these disorders emerged in a recent study, led by Harris, of a cellular suicide switch known as Bax. Harris had read about experiments from other researchers linking Bax to nerve cell suicide in other neurodegenerative disorders, so he decided to see what would happen in one of his mouse models if the Bax gene was knocked out. As he had hoped, the alteration saved a class of mouse brain cells normally killed off in dramatic fashion in the mouse model of the prion disorder. But the mice still developed movement disorders and other symptoms that were characteristic of their condition when they had a functioning Bax gene. Further investigation revealed extensive damage to the synapses, areas where branches of two brain cells come together to communicate. “ This connects prion diseases to other more common disorders because it shows nerve cell death isn’t the only thing we have to worry about in these conditions,” Harris explains. “We have to be concerned about damage to the synapse too, and there’s increasing evidence that is the case in other disorders like Alzheimer’s disease.” That may make a big difference for therapeutics currently in development, Harris notes. “ Our results suggest that if we just prevent cell death without doing something to maintain the functionality of the synapse, patients may still get sick,” he says. Although they work on very different aspects of prion research, True-Krob and Harris collaborate on projects, have a monthly joint lab meeting, and interact frequently. Harris jokes that he and True-Krob make up “the largest center of prion research within 1,000 miles or so.” True-Krob notes when she was looking for her first faculty position, the possibility of coming to a department with another faculty member studying prions had “definite appeal.” Their field may soon be getting much less lonely. Connections to more common neurodegenerative disorders are increasing, and other researchers (including True-Krob’s postdoctoral mentor) recently proposed that prions may help store memories in brain cells. “
That theory’s got a long way to go,” True-Krob says, “but
it’s
indicative of a new willingness to think about the possibility
that prions could have a beneficial role in other systems besides yeast.
More and more people are
becoming aware of the prion and considering it as a possible explanation
for puzzling results.”
|
||||||