

-

- Oasis
- Scratching the surface
- Critical differences
- Safer passage

Critical differences count
A highly efficient — and effective — method for finding disease-related genes takes a shortcut via complex math
BY Caroline Arbanas
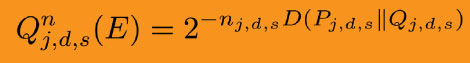
Adapting Sanov’s theorem resulted in a more efficient variation-finder.
A major motivation behind the $3 billion project to decode the human genome was that it would enable scientists to sift through the sequence of letters that make up DNA to find common changes that predispose individuals to common diseases, such as cancer, diabetes or Alzheimer’s.
But this first step toward personalized genomic medicine has turned out to be far more complex than expected.
Many genetic studies have compared common variations in the DNA of healthy people and sick patients, finding dozens of changes linked to diseases. But variations identified to date account for only a small percentage — typically 1 percent to 3 percent — of overall genetic risk for any particular disease.
That disappointment has led a growing number of scientists to suspect that rare genetic variants lie at the root of many common diseases. But short of sequencing the complete genomes of many thousands of individuals — a highly expensive and time-consuming task even with the latest DNA sequencing technology — no reliable method exists to identify rare variants or interpret their influence on disease.
“There’s intense interest in rare variants right now,” says pediatric oncologist Todd E. Druley, MD, PhD. “We couldn’t wait until whole-genome sequencing became cost effective before we started our search.”

Todd E. Druley, MD, PhD, Robi D. Mitra, PhD, and Francesco Vallania
The urgency hits home every time Druley has to tell parents their child has cancer. “Of course, the first thing parents want to know is whether their child will die. Then, invariably, they ask: ‘Why did this happen to my child?’”
Not knowing the answer was the driving force behind Druley’s efforts to find a way to search for rare variants. Toward that end, he teamed with Robi D. Mitra, PhD, assistant professor of genetics, who specializes in the development of experimental and computational tools that allow biologists to collect large volumes of genetic data.
“We think children with cancer are likely to have a unique collection of rare, inherited DNA variations that predispose them to their disease or to difficulties metabolizing the medications used to treat cancer,” Mitra says. “Rare variants likely contribute to disease in adults, too, but until now, we haven’t even had a way to investigate the link.”
With funding from the Children’s Discovery Institute, the team developed a way to find and quantify all the inherited rare genetic variants in a complete set of human genes. Their method is fast and inexpensive — and incredibly accurate, they reported in the April 2009 issue of Nature Methods.
The approach, which combines next-generation DNA sequencing technology with a computer program the research team developed, has attracted the attention of other sci-entists. The team now is working with the Children’s Oncology Group, the global clinical trials cooperative, to identify rare variants in pediatric cancer patients with an aggressive form of leukemia.
The bulk of the 3 billion pairs of chemical bases that make up DNA — represented by the letters A, T, C and G — are identical from one person to the next. But the exact order of the letters varies, often at a single location, in some 10 million common spots in the genome. These variations are what make each individual unique.

Each person also has about 300,000 rare genetic alterations buried within the genome. These variations mean nothing by themselves; it’s only in the context of a large group that scientists can identify their links to a particular disease.
To find individual DNA samples to search for rare variants, Druley and Mitra turned to F. Sessions Cole, MD, the Park J. White MD Professor of Pediatrics and director of the Division of Newborn Medicine, who provided his collection of thousands of blood samples collected from the heels of newborns.
The researchers pooled the DNA of 1,111 newborns and targeted four genes. To ensure accurate results, they created thousands of copies of the four genes and sequenced them many times to identify the full spectrum of genetic variation.
The genes of interest included surfactant protein B, which Cole discovered in 1993 and linked to respiratory distress syndrome in infants. Cole had painstakingly sequenced the gene in all 1,111 individuals with traditional sequencing methods; Mitra and Druley used the gene as a control.
“We wondered whether we would find the same variants at the same frequencies in our pooled sample that Dr. Cole had already identified,” Mitra says.
They also looked for rare variants in two genes important in cancer — p53 and APC — as well as in beta actin. None of these three genes had been sequenced in the 1,111 individuals.
In all, the team sifted through a haystack of 4.5 billion DNA bases. They found 64 variants in the four genes, nearly two-thirds of which were identified in less than 1 percent of the individuals, making them rare. These included four variants that had not been previously identified. Three of them occurred in the two cancer genes.
The computer algorithm, developed in collaboration with graduate student Francesco Vallania, accurately distinguished between rare variants and sequencing errors most of the time. “This is crucial because rare variants can easily look like sequencing errors,” Mitra says. “How do you tell the difference? The algorithm we developed could do that.”
The researchers accurately recapitulated all of Cole’s data, finding variants at the predicted frequencies in the surfactant protein B gene. They also went back to verify the frequencies of variants in the other three genes. In all, they accurately identified all the rare variants in the pooled sample. They also identified sequencing errors as mistakes 99.8 percent of the time. Overall, the algorithm was 96 percent accurate.
With the new method, the sequencing that took Cole 2.5 years at a cost of $200,000 was completed in just four months for $6,000.

“This provides proof of principle,” Druley says. “We’re now testing the approach in children with aggressive leukemia and in healthy children to see whether we can find rare variants linked to the disease.”
Ideally, the team hopes to identify rare variants linked to prognosis that could be evaluated in newly diagnosed patients. These variants may tell doctors which patients need aggressive therapy or which treatment works best in a particular patient.
Since the original publication, they have expanded their approach by adding a DNA barcode onto each individual’s contribution to the pooled DNA sample. This enables them to trace rare variants back to individual patients, which was not previously possible.
The investigators also are collaborating with other Washington University researchers by using the method to find rare variants in other complex diseases: newborns with lung disease, children with clubfoot, brain tumors or sickle cell disease, adults with heart failure and lung disease.
“Ultimately, we think we’ll be able to use this simple method to find important genetic variations,” Mitra says. “Lots of people have done genetic studies and have come up empty-handed. They’re anxious to get at rare variants, and we think this method will get us there, not just for cancer but for many important diseases.”
